Description
Introduction
The success for designing high efficiency engines largely depends
on our ability to process materials that can withstand high temperatures
without failure. A tremendous amount of research has been devoted
to finding new Ni-based superalloys which are thermodynamically
and structurally stable at higher temperatures and for longer
periods of time. Ni-based superalloys consist of ordered intermetallic
g'(Ni3Al) precipitates embedded in a disordered face-centered
cubic (fcc) g matrix. The control of the g+ g' two-phase microstructure
and its high-temperature stability is the key to the success for
the development of superalloys with desired high temperature properties
since the g' precipitate volume fraction, precipitate morphology,
and distribution in these alloys strongly affect their mechanical
properties (e.g. strength, fatigue and creep). The precipitate
morphology and spatial distribution are known to depend on a number
of factors which include the precipitate volume fraction, alloy
compositions, temperature, lattice mismatch between the precipitates
and matrix, presence of dislocations, and applied stress loads.
In current laboratory and industrial practice, the traditional
trial-and-error method is still the major approach to optimizinge
the alloy chemistry and processing conditions for achieving desirable
desired microstructuresmechanical properties for of Ni-based superalloys.
This usually requires many long and expensive experimentations.
However, with the rapid development in computer technology and
recent advances in computational modeling, we believe it is now
possible to develop a set of physics-based, user-friendly computational
tools that can be employed for both fundamental materials research
and design of new advanced materials. The main objective of the
proposal is to develop a set of integrated and predictive computational
tools that can be employed to determine the relationships among
chemistry, microstructure and mechanical properties of single-crystal
superalloys at high temperatures ( > ).
Proposed approaches, the rationale, and new innovations
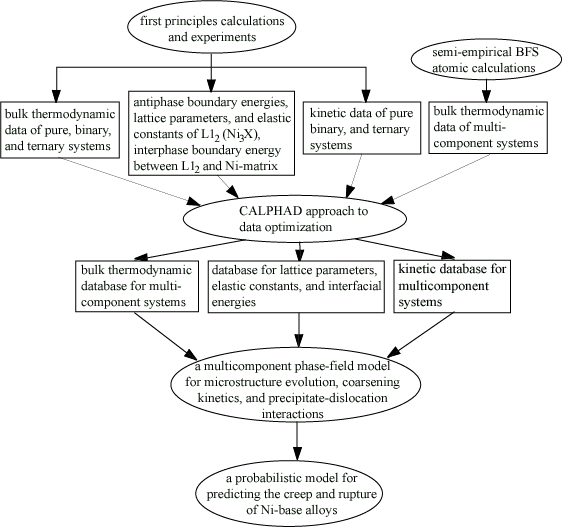
We proposed to build the set of computational tools based on the
following approaches (See Fig. 1): (ia) first-principles calculations
of the thermodynamic and kinetic parameters, lattice parameters,
and elastic constants for low-order systems including single-component
systems, binary and ternary alloys as well as thermodynamic properties
of point defects, dislocations and antiphase boundary energies
in such systems, and semi-empirical atomistic calculations of
thermodynamic properties of multicomponent systems using the BCS
(Bozzolo-Ferrante-Smith) method[1];, (iib) computational thermodynamicsCALPHAD
(often referred to as CALculation of PHAse Diagrams) approach[2]
for to constructing thermodynamic, kinetic and crystallographic
databases for multicomponent systems using the data obtained from
first-principles calculations, semi-empirical BCS method, and
experimental measurements in low-order systems (number of components
> 3) based on the thermodynamic and kinetic data obtained from
low order systems and for determining the phase-equilibria and
thermodynamics of multicomponent alloys; (iiic) a phase-field
method[3] for predicting the microstructure evolution and coarsening
kinetics of g' gamma-prime precipitates in multicomponent alloys
with the fundamental thermodynamic, kinetic and crystallographic
input directly from databases with input from the thermodynamic
and kinetics databases; and (ivd) a ?? creep property method ofmodel
for predicting and extrapolating approach for modeling the creep
and rupture properties of single-crystal Ni-based superalloys
with information obtained from the microstructure model, crystallographic
databases, and creep measurements using the microstructure information
obtained from phase-field simulations. For validating the computational
models, critical experiments on the determination of thermodynamic
data and high-temperature phase-equilibria will be conducted at
Penn State using the newly developed combinatorial approach. The
measurement of coarsening kinetics of g' gamma-prime precipitates
at high temperatures (> precipitates will also be performed
at Penn State in collaboration with the Advanced Metals Branch
of the Materials Division at NASA-GRC, and high-temperature creep
testing of single-crystal Ni-based superalloys will be conducted
at University of Florida. All the experimental validation will
be performed in close collaboration with the Advanced Metals Branch
of the Materials Division at NASA-GRC. In addition, Dr. Zhao at
General Electric, who pioneered the combinatorial approach for
structural materials[4, 5], has agreed to help and advise a graduate
student in carrying out part of the experimental validation of
thermodynamic/kinetic databases.
The rationales for proposing such a multiscale computational
approach are as follows. Recent advances in first-principles calculations
have made it possible to predict accurate thermodynamic properties,
such as formation energies and enthalpies, of pure and binary
alloys, ternary and multicomponent alloys using only the atomic
numbers as the input. However, up to now it is not computationally
tractable, for the foreseeable future, to use first-principles
calculations to accurately determine the configurational and vibrational
entropy contribution to the total free energy directly from first-principles
for a multicomponent system. Several studies have shown progress
recently in binary systems. rationalsOn the other hand, semi-empirical
methods based on the CALPHAD approach have been very successful
in determining the phase equilibria of multicomponent commercial
alloys, often with ten components or more[17, 18]. The CALPHAD
approach builds thermdynamicthermodynamic databases for multicomponent
systems using data obtained in single-component, binary and ternary
systems. Therefore, a marriage of first-principles calculations
of simple low-order systems and the CALPHAD approach will allow
one to develop thermodynamic databases for multicomponent systems
from first-principles. Since the CALPHAD approach uses extrapolation
of the data from experiments and first-principles calculations
of low-order systems to multicomponent systems, its accuracy for
multicomponent systems must be validated experimentally. To increase
the database reliability, we plan to utilize the expertise at
NASA on semi-empirical atomic calculations using the BFS method
to obtain additional thermodynamic data, in particular, the formation
enthalpies, of multicomponent systems in the database assessment.
A similar strategy can be adopted for developing kinetic databases
and databases for lattice parameters, elastic constants and interfacial
energies as a function of composition and temperature.
Fig. 1. An integrated set of computational tools
A recent important development in computational materials science
is the emergence of the powerful phase-field approach for modeling
phase transformations and microstructure evolution. It is based
on the fundamental thermodynamic and kinetic principles, but it,
which requires input data for the thermodynamic and kinetic parameters,
the lattice parameter and elastic constants dependence on composition
and order parameters. One of the main advantages of the field
approach is that the temporal evolution of any arbitrary microstructures
can be predicted without any a priori assumptions about their
evolution path. Precipitate disappearance and coalescence are
automatically taken into account, so there is no limit on the
volume fractions of precipitates that can be modeled. There is
no technical difficulty in extending from 2D to 3D simulations
except a significant increase in computational time and memory
requirements. We recently showed that even with current computer
technology, we were able to model realistic 3D systems with thousands
of g' precipitates in a g matrix at any given volume fractions[6].
On the other hand, using the traditional front-tracking models,
even the state-of-the-art boundary-integral method[7] is only
able to model the coarsening kinetics of precipitates at low volume
fractions; it cannot handle the topological changes during coalescence
of precipitates at high volume fractions, even in two dimensions
(2D), while the volume fractions of g' precipitates in most Ni-base
superalloys are typically about 50% or higher[8]. Using front-tracking
sharp-interface models, it is technically impossible to model
the evolution of more realistic 3D microstructures with high volume
fraction of precipitates, although modeling coarsening of small
volume fractions of spherical precipitates is possible. Moreover,
a traditional front-tracking model cannot distinguish the four
types of antiphase-related domains of g' precipitates that coexist
in a g+g' two-phase microstructure, which can have a significant
influence on the precipitate morphology and coarsening kinetics.
The difficulty of a sharp-interface description in treating the
topological changes during microstructure evolution is also the
main reason that the commercial code "DICTRA" can only be used
to model one-dimensional (1D) phase transformation and microstructure
problems. DICTRA is a general software package for simulation
of Diffusion Controlled TRAansformations in multicomponent systems
developed by the Royal Institute of Technology in Sweden. If the
proposed project is funded and successfully completed, a potentially
much more sophisticated computational tool than the current commercial
code "DICTRA" can be developed. Coupled with reliable thermodynamic
and kinetic databases, the proposed model can be used to predict
the coarsening kinetics of g' precipitates in multicomponent single-crystal
Ni-base superalloys with only the temperature and compositions
as the input, and therefore can be used in the design of superalloy
microstructures.
Our ultimate interest is to predict the mechanical properties
of an multicomponent alloy as a function of alloy chemistry, temperature
and time. Therefore, it is critical to link microstructures predicted
from phase-field simulations to the tensile and creep behavior
of Ni-based superalloys. For the tensile properties at relatively
low temperatures, the recent breakthrough advances in phase-modeling
of dislocation microstructures and dislocation slipping dynamics
makes it possible to study the effect of alloy chemistry, and
thus various solute species and the g' precipitate size, morphology,
and volume fraction on the stress-strain curve of Ni-base alloys.
However, for high temperatures at which multiple slip, cross-slip,
and dislocation climb are taking place, significant additional
effort is required to make the current phase-field technology
directly applicable to high-temperature creep. Moreover, in real
commercial superalloys, metallurgical defects (e.g., porosity,
inclusions, low-angle boundaries) due to processing are common.
To accurately predict the creep and the lifetime of an engine
component, the effect of these randomly created processing defects
on the creep properties must be incorporated in a creep model,
in addition to the effects of microstructural details (e.g., precipitate
size and morphology) which can be obtained from the microstructure
model. Therefore, one of the goals of the proposed research is
to develop a model that can predict the properties of Ni-base
superalloy single crystals that combines a probabilistic creep
property parameter. To develop such a model, extensive laboratory
testing of model and commercial alloys will be used to quantify
the effects of microstructural details, alloying additions and
metallurgical defects on the creep properties.
The proposed computational tools are highly innovative in the
following aspects. First of all, although the CALPHAD approach
has existed for more than two decades and has been extensively
used in calculating the phase equilibria in complex alloys, as
far as we are aware of, the proposed research presents the first
systematic effort to construct thermodynamic and kinetic databases
by using extensively first-principles and semi-empirical atomistic
calculations. Such an approach will allow one to build databases
which can be used to predict the phase equilibria in experimentally
unexplored compositional fields and for alloying species which
have not been studied experimentally. It will also improve the
reliability of databases by incorporating the thermodynamic data
of certain metastable or stable precipitates that are difficult
to isolate experimentally and whose structure and thermodynamic
properties are almost impossible to measure experimentally. Secondly,
the proposed research is the first attempt to build a database
for the lattice parameters (crystallographic), elastic constants,
and interfacial energies as a function of composition and temperature
for complex alloys. Third, preliminary phase-field models for
simple, elastically homogeneous binary Ni-base superalloys have
been developed at Penn State and other universities during the
last few years. The proposed effort in microstructure modeling
will be focused on developing an innovative computational microstructure
model for complex, elastically inhomogeneous, multicomponent systems
by incorporating recent advances at Penn State in developing fast
numerical algorithms for solving phase-field equations, capability
for modeling elastically inhomogeneous systems under applied stresses,
and a novel formulation for modeling precipitate microstructure
evolution in the presence of dislocations. Such an innovation
is critical for modeling the microstructure evolution in real
alloys in which the elastic constants are always inhomogeneous
and most likely multicomponent and in alloys under an applied
stress. Fourth, the proposed creation of an interface between
thermodynamic, kinetic, and crystallographic databases and the
phase-field model is a first major step to realize the potential
application of the phase-field model to practical alloy designs.
Finally, we proposed the first major effort to build a constitutive
creep model with input from a microstructure model while incorporating
the effect of processing defects such as porosity and low-angle
boundaries. The proposed probabilistic model will be a first of
its kind and it promises to improve significantly the accuracy
of existing extrapolation/interpolation model.
|

